
 Le dépistage néonatal permet de détecter et de prendre en charge de manière précoce, chez tous les nouveaux-nés, des maladies rares, sévères et le plus souvent génétiques ; il est pris en charge à 100 % par l’Assurance maladie. Couramment appelé « test de Guthrie », ce dépistage permet de rechercher la présence de 13 maladies. Désormais, le dépistage de la drépanocytose n’est plus seulement réservé aux enfants présentant un risque particulier de développer la maladie.
Le dépistage néonatal permet de détecter et de prendre en charge de manière précoce, chez tous les nouveaux-nés, des maladies rares, sévères et le plus souvent génétiques ; il est pris en charge à 100 % par l’Assurance maladie. Couramment appelé « test de Guthrie », ce dépistage permet de rechercher la présence de 13 maladies. Désormais, le dépistage de la drépanocytose n’est plus seulement réservé aux enfants présentant un risque particulier de développer la maladie.
Le programme de dépistage néonatal existe depuis 1972. Il se caractérise par des examens de biologie médicale ; ceux-ci sont assurés dans chaque région par un centre régional de dépistage néonatal (CRDN) rattaché à un centre hospitalier universitaire (CHU) en lien avec les agences régionales de santé (ARS).
Le dépistage néonatal est réalisé en prélevant des gouttes de sang sur un buvard, après une petite piqûre au talon du nouveau-né. Il est systématiquement proposé et réalisé après accord des parents. Le prélèvement est fait le plus souvent en maternité, parfois au domicile, dans les 3 jours suivant la naissance. Les résultats ne sont communiqués aux parents qu’en cas de problème.
À la suite des recommandations de la Haute Autorité de santé, la drépanocytose, maladie génétique la plus fréquente parmi celles décelées à la naissance, va être dépistée systématiquement chez tous les nouveau-nés, c’est ce qu’indique un arrêté publié au Journal officiel du 3 août 2024. Auparavant son dépistage était soumis à conditions.
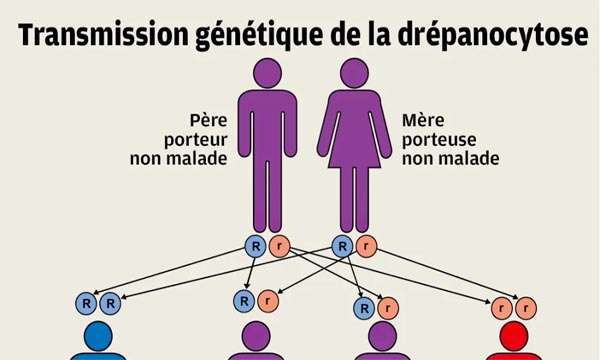
Qu’est-ce que la drépanocytose et quels sont les symptômes ?
La drépanocytose, aussi appelée anémie falciforme, est une maladie génétique héréditaire touchant les globules rouges (ou hématies). Elle est caractérisée par une anomalie de l’hémoglobine, principale protéine du globule rouge.
Les symptômes de la maladie sont variables et dépendent non seulement de l’âge, mais aussi de la sévérité de la drépanocytose.
Dans les tous premiers mois, les nourrissons sont généralement asymptomatiques car ils bénéficient de la présence d’hémoglobine fœtale, la forme d’hémoglobine produite chez le fœtus durant la période in utero, qui n’est pas mutée quant à elle. Les premières complications surviennent à partir de 3 mois environ avec des crises douloureuses et une anémie aiguë pouvant causer essoufflement et fatigue. Cette maladie est également responsable d’une sensibilité accrue aux infections bactériennes (pneumonies, méningites ou septicémies…).
Chez les tout-petits, la crise se manifeste généralement par un gonflement douloureux des mains et des pieds (syndrome pied-main).
À noter
la drépanocytose est présente partout dans le monde, mais touche préférentiellement certaines régions : Afrique, Bassin méditerranéen, Asie. Le dépistage de la drépanocytose se généralise à l’ensemble des nouveau-nés du territoire depuis le 4 août 2024.
Quelles sont les 12 autres maladies graves de l’enfant actuellement dépistées ?
Les maladies recherchées par le dépistage sont les suivantes :
- la phénylcétonurie : maladie génétique due au déficit d’une enzyme qui transforme la phénylalanine présente dans l’alimentation. En l’absence de traitement, elle peut entraîner un retard mental sévère et des complications neuropsychiatriques ;
- l’hypothyroïdie congénitale : maladie qui se traduit par une sécrétion insuffisante des hormones thyroïdiennes par la glande thyroïde. En l’absence de traitement, son dysfonctionnement retentit sur les grandes fonctions de l’organisme et peut avoir notamment pour conséquence, un retard mental sévère ;
- l’hyperplasie congénitale des surrénales : défaut génétique du fonctionnement des glandes surrénales. En l’absence de traitement, elle peut être à l’origine de déshydratations aiguës sévères, parfois mortelles, et de troubles du développement des organes sexuels ;
- la mucoviscidose : maladie génétique qui entraîne des infections respiratoires sévères et répétées ainsi que des complications digestives. Une prise en charge précoce permet de mettre en place des procédures pour assurer un bon état nutritionnel de l’enfant, faciliter l’évacuation des sécrétions bronchiques, prévenir les infections et les traiter ;
- le déficit en MCAD (Medium-Chain-Acyl-CoA Déshydrogènase) : maladie qui entraîne une difficulté de l’organisme à utiliser les graisses comme source d’énergie. En l’absence de traitement, elle peut provoquer des comas pouvant aller jusqu’au décès de l’enfant ;
- l’homocystinurie : maladie génétique liée au déficit d’une enzyme, la « cystathionine bêta-synthase », qui entraîne l’accumulation d’homocystéine toxique pour l’organisme. En l’absence de traitement, elle peut entraîner une atteinte des yeux, du squelette, du système vasculaire, du système nerveux et parfois un retard du développement ;
- la leucinose ou maladie des urines à « odeur de sirop d’érable » : maladie génétique liée au déficit d’une enzyme, la « déshydrogénase des alpha-céto-acides à chaîne ramifiée ». En l’absence de traitement, elle se caractérise par l’apparition rapide de difficultés pour s’alimenter, un temps de sommeil trop prolongé, des vomissements puis des troubles neurologiques, des mouvements anormaux et une insuffisance respiratoire
- la tyrosinémie de type 1 : maladie génétique liée au déficit de l’enzyme hépatique, la « fumaryl acétoacétate-hydrolase » qui permet la transformation normale des protéines contenues dans les aliments. Sans un régime et un traitement appropriés, des déchets toxiques s’accumulent dans l’organisme et endommagent gravement le foie, les reins et le système nerveux ;
- l’acidurie isovalérique : maladie génétique, appelée également acidémie isovalérique, liée au déficit d’une enzyme, « l’isovaléryl-CoA déshydrogénase », responsable en l’absence d’un régime adapté de troubles aigus à la naissance (vomissements, convulsions) ou plus tardifs (retard de croissance et/ou de développement) ;
- l’acidurie glutarique de type 1 : maladie génétique liée à l’absence ou l’insuffisance de fonctionnement d’une enzyme, « la glutaryl CoA-déshydrogenase ». En l’absence d’un régime spécial, elle est à l’origine de l’accumulation de produits toxiques responsables de troubles neurologiques aigus chez les nourrissons ;
- le déficit en 3-hydroxyacyl-coenzyme A déshydrogénase des acides gras à chaîne longue : maladie génétique liée à l’absence ou l’insuffisance de fonctionnement de cette enzyme. En l’absence de traitement et d’un régime adapté, elle se caractérise par la survenue dans la petite enfance d’une hypoglycémie, d’une atteinte au foie, de troubles cardiaques et neurologiques ;
- le déficit primaire en carnitine : maladie génétique liée au déficit de transporteur de la carnitine. En l’absence d’administration de carnitine, ce déficit entraîne une atteinte cardiaque au début de l’enfance, souvent associée à une hypotonie, un retard de croissance, des crises hypoglycémiques récurrentes et/ou un coma.
Le nombre de maladies dépistées est susceptible d’augmenter en fonction des avis rendus par la Haute Autorité de santé sur les nouveaux dépistages.
Le programme est complété par le dépistage de la surdité permanente néonatale.
Le dépistage des 13 maladies rares s’ajoute aux pathologies déjà dépistées.
Voir aussi
- J’attends un enfant
Service-Public.fr
- Calendrier des vaccinations
Service-Public.fr
- Calendrier vaccinal : quels changements pour 2024 ?
Service-Public.fr
- 1 000 premiers jours : s’informer pour l’arrivée d’un enfant
Service-Public.fr
- Détecter et prendre en charge précocement 13 maladies graves de l’enfant
Ministère chargé de la santé
- Ce que vous devez savoir sur la drépanocytose
Ministère chargé de la santé
- Suivi de l’enfant à la maternité
Caisse nationale d’assurance maladie (Cnam)
Publié le 06 août 2024 – Direction de l’information légale et administrative (Premier ministre)